
Abstract
Introduction
Biologic treatments are increasingly being used in the management of moderate to severe plaque psoriasis (PSO). Bimekizumab is a selective inhibitor of both interleukin (IL)-17A and IL-17F approved for the treatment of moderate to severe PSO. Although bimekizumab trials provide comparisons to secukinumab, adalimumab and ustekinumab, there are no further head-to-head comparisons of bimekizumab to other biologics. This network meta-analysis (NMA) aimed to compare the short-term efficacy of bimekizumab versus other biologic systemic therapies for moderate to severe PSO.
Methods
A systematic literature review was conducted to identify randomised controlled trials (RCTs) in patients with moderate to severe PSO. MEDLINE, Embase, the Cochrane Central Register of Controlled Trials and the Database of Systematic Reviews and PsycINFO were searched on July 1, 2020. An enhanced multinomial Bayesian NMA model was used to evaluate the comparative efficacy in 50%, 75%, 90% and 100% improvement from baseline Psoriasis Area and Severity Index (PASI 50/75/90/100) at 10–16 weeks. The model was also adjusted for baseline risk, given the variable placebo responses across the trials.
Results
Eighty-six RCTs (including 34,476 patients) were included in the NMA. IL-17 and IL-23 inhibitors were the most effective treatments across all PASI levels. At 10–16 weeks, bimekizumab had the highest probability of achieving PASI 75 (92.3%), PASI 90 (84.0%) and PASI 100 (57.8%). Bimekizumab demonstrated statistical superiority over all biologics in achieving PASI 90 and PASI 100 thresholds. For PASI 75, the benefit of bimekizumab was statistically significant compared to all other treatments except risankizumab and ixekizumab.
Conclusion
This analysis demonstrated that IL-17 and IL-23 inhibitors were highly effective in achieving short-term improvement among patients with moderate to severe PSO. Patients receiving bimekizumab were significantly more likely to achieve PASI 90 or PASI 100 within 10–16 weeks of the first injection than all other biologics.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Why carry out this study? |
Plaque psoriasis (PSO) is an immune-mediated disease associated with increased mortality, substantial impact on quality of life and major economic burden to health systems |
Over the past decade, several biologic therapies have been introduced for the treatment of moderate to severe psoriasis, which led to a transformational impact on treatment outcomes. Nevertheless, the evidence on the role of biologic treatments in the management of moderate to severe PSO is largely based on placebo-controlled randomised controlled trials with limited head-to-head comparisons |
What did the study ask?/What was the hypothesis of the study? |
This systematic literature review and network meta-analysis (NMA) evaluated the comparative efficacy of bimekizumab and other approved biologic therapies in achieving skin clearance (as examined using the Psoriasis Area and Severity Index) within 10 to 16 weeks of treatment |
What were the study outcomes/conclusions? (data) |
At 10 to 16 weeks, the NMA (k = 86) demonstrated that interleukin (IL)-17 and IL-23 inhibitors were more effective treatments than tumour necrosis factor inhibitors across all PASI response levels with bimekizumab 320 mg displaying the highest probability of achieving PASI 75, PASI 90 and PASI 100 |
What has been learned from the study? |
Bimekizumab demonstrated statistical superiority over all biologics in achieving PASI 90 and PASI 100 thresholds, whereas for PASI 75, the benefit of bimekizumab was statistically significant compared to all other treatments except risankizumab and ixekizumab |
Introduction
Plaque psoriasis (PSO) is an immune-mediated disease characterised by flaky, scaly, itchy and red skin plaques [1] and accounts for up to 90% of psoriasis cases [2]. PSO affects approximately 1.5% to 2% of the general population of Western Europe and North America with incidence rates peaking in the 4th decade of life [3]. Severity of PSO depends on location, symptom profile and intensity, surface area involved and impact on daily functioning [4]. About 20% of patients with PSO have moderate to severe disease, which is commonly defined as involving > 10% of the body or affecting crucial body parts [2, 5]. Severe disease is associated with increased mortality, substantial impact on quality of life and major economic burden to health systems [6, 7].
Conventional therapies, including systemic non-biologic drugs such as methotrexate and cyclosporine, are widely used and recommended by several practice guidelines including the Joint American Academy of Dermatology and the British Association of Dermatologists guidelines for the management of psoriasis [8, 9]; however, their efficacy is limited in patients with moderate to severe PSO, and they are associated with a risk of major long-term toxicity [10]. Biologic therapies have revolutionised the management of PSO, offering highly effective and tolerable treatment options to healthcare providers and patients. They are generally recommended for patients who have a total Psoriasis Area and Severity Index (PASI) score ≥ 10, a Dermatology Life Quality Index (DLQI) score > 10 and are resistant to treatment with traditional systematic drugs based on intolerance, contraindications or failure in response [11]. Currently approved groups of biologic therapies for PSO include interleukin (IL) antagonists and tumour necrosis factor (TNF)-α targeting agents. IL antagonists target pro-inflammatory cytokines, including the IL-12/23p40 antibody (ustekinumab), and, more recently, inhibitors of IL-17A (secukinumab and ixekizumab), IL-17RA (brodalumab) and IL-23p19 (guselkumab, tildrakizumab and risankizumab) [12]. Bimekizumab, presently approved in Australia, Canada, the European Union, Japan and the UK [13,14,15,16,17], is the only approved selective inhibitor of both IL-17A and IL-17F. IL-17F is abundant in skin lesions and can drive inflammation independently of IL-17A [18]. Bimekizumab prevents these cytokines from binding to their cellular targets, inhibiting them from promoting inflammation, and thus reducing the symptoms of PSO. With its novel dual inhibition mechanism of IL-17A and IL-17F, bimekizumab has recently been reported to offer a rapid and durable skin clearance in patients with moderate to severe PSO [19,20,21,22].
Evidence on the role of biologic treatments in the management of moderate to severe PSO is largely based on placebo-controlled randomised controlled trials (RCTs). Although bimekizumab trials (BE RADIANT, BE SURE, BE VIVID) [20,21,22] provide head-to-head comparisons to secukinumab, adalimumab and ustekinumab, there are no direct comparisons of bimekizumab to other biologics. Indirect or mixed treatment comparisons are necessary for informing decisions about treatment choices by clinicians and patients. The objective of this network meta-analysis (NMA) was to compare the efficacy of bimekizumab and other approved biologic systemic therapies for moderate to severe PSO. The analysis focused on the efficacy data at the end of induction treatment (10 to 16 weeks)—specifically, the proportions of patients achieving commonly reported percentage changes with the PASI relative to baseline.
Methods
Systematic Literature Review
This systematic literature review (SLR) was conducted in accordance with the Cochrane Collaboration [23] and the Preferred Reporting Items for Systematic Reviews and Meta-Analyses [24] guidelines to identify RCTs assessing the short-term efficacy and safety of biologic and non-biologic therapies in the management of moderate to severe PSO. Searches of MEDLINE, Embase, the Cochrane Central Register of Controlled Trials, the Cochrane Database of Systematic Reviews, and PsycINFO were conducted on March 5, 2019, and updated on July 1, 2020, to identify English-language studies conducted on humans. Database searches were conducted using search terms and keywords for moderate to severe PSO, approved treatments for the disease and study design of interest (i.e., RCTs) (Table S1 to Table S4 in the electronic Supplementary Material), and were supplemented by a review of proceedings from seven PSO-related conferences published between 2016 to 2020 . Bibliographies of relevant SLRs identified in the search (published between 2016 and 2020) were manually examined for any studies not identified by the searches.
Data from five bimekizumab trials were included in the SLR. These included the published phase 2b (BE ABLE 1 [19]) and four phase 3/3b trials that were not yet published at the time of the review (data on file for BE READY, BE SURE, BE VIVID and BE RADIANT were made available [25,26,27,28]).
Study Selection
Articles identified through database searching were screened against the pre-defined inclusion/exclusion criteria, defined by the population, interventions, comparators, outcomes, study design, and time framework (Table S5 in the electronic Supplementary Material). Articles were included if they reported on RCTs investigating the efficacy (assessed via percentage improvement in the PASI from baseline) of biologic therapies (at dosages approved by the European Medicines Agency) and non-biologic therapies at the end of the induction treatment phase (10 to 16 weeks) for adults with moderate to severe PSO. Phase 2 trials were considered for inclusion only if two treatments arms were evaluated (i.e., a licensed dose strength of a biologic intervention of interest and placebo); additional treatment arms of unlicensed dose strengths were excluded from the evidence base. Title and abstract screening and full-text review were conducted independently by two reviewers. Discrepancies were resolved by consensus or by a third, more senior reviewer.
Data Extraction and Risk-of-Bias Assessment
Data from the included studies were extracted by one researcher into pre-designed standardised data extraction forms. Data elements included study characteristics and patient characteristics (including demographic characteristics, comorbidities, disease duration and prior treatment), treatment details and outcomes of interest for each RCT. All extractions were independently validated by a second investigator. The quality of all RCTs was assessed using the Cochrane Risk of Bias Assessment Tool 2.0 [29].
When more than one publication was identified for the same RCT, data extracted from the primary publication were supplemented with more recent data available in related publications.
NMA Assumptions
NMAs are based on the assumption that the underlying relative treatment effects (between any two specific treatments, after disregarding the sampling error) are the same in all trials [30]. This study assessed the presence of potential effect modifiers [31], such as disease duration, baseline PASI scores, prior biologic therapy use, and presence of comorbidities to confirm consistency and similarity among the eligible trials that connected to the network. Since placebo response may also interact with relative treatment effect, these differences were examined and are presented in Figure S1 in the electronic Supplementary Material. Small differences in treatment doses and schedule in the non-biologic treatments cyclosporine and methotrexate were assumed to have no impact on relative effects; this assumption was based on expert clinical review and allowed for network connectivity.
NMA
A Bayesian multinomial likelihood NMA model (probit link) was conducted to compare the relative effects for 50%, 75%, 90% or 100% improvement from baseline (PASI 50/75/90/100) across treatments at 10 to 16 weeks. This time frame encompasses the range of stated primary end point time points across studies, and we selected PASI findings at the stated primary end point for each of the studies included in the NMA. This study explored clinical heterogeneity and the performance of NMA models using unadjusted and adjusted models per the National Institute for Health and Care Excellence (NICE) Decision Support Unit recommendations [32,33,34]. This NMA focused on efficacy outcomes, and safety data were not incorporated.
The NMA was based on the NICE NMA model for standard multinomial analysis [34] including a component for baseline risk. Baseline risk was considered as the relative effects of drugs in autoimmune diseases, particularly for treatment response, may depend on baseline risk (i.e., the placebo rate and relative effect of a treatment versus placebo are often related [34]).
Furthermore, because the standard multinomial model assumes that a relative treatment effect (i.e., probit difference) is identical for all PASI levels, we modified the model to allow the relative treatment effects to vary across PASI thresholds. This modification, introduced by Fahrbach et al. in another NMA of moderate to severe PSO [35], added a random-effects (RE) component to the parameter z and is referred to below as the ‘REZ’ model. This addition models each treatments’ increase in difficulty to the next-highest PASI cut-off as varying around a common mean while the standard model assumes all treatments have exactly the same mean difficulty (in probit terms) in achieving a higher PASI cut-off. The practical impact of this model addition is that it allows treatment rankings to differ across different PASI cut-offs, which is not possible under the standard model.
All analyses were run with fixed-effects (FE) and RE modelling for relative treatment effects. Eight different analytic scenarios were tested by crossing FE/RE for treatment effects with the aforementioned modelling modifications (i.e., the inclusion/exclusion of baseline risk and the use of standard versus REZ modelling). Binomial analyses with a logit link for all PASI responses were performed as sensitivity analyses; however, some interventions were not compared because of lack of trial data. In addition, the rarity of events for PASI 100, particularly in the placebo arms of numerous trials, precluded the conduct of the binomial sensitivity analysis for this threshold.
The posterior mean residual deviance and deviance information criteria (DIC) were used to compare the goodness of fit of the eight analytic multinomial models (i.e., a better-fitting model is generally one with a DIC smaller by > 5 points) [34, 36]. Other considerations for the selection of the base case model included the current understanding of PSO (i.e., relative effects depend on placebo rates) [31], practice of the analytic approaches, expert opinion and data availability.
A Bayesian NMA was conducted (see the appendix in the electronic Supplementary Material for detailed description of the prior choice, Monte-Carlo simulations and model convergence assessment) from which the median and (2.5th and 97.5th) percentiles of the posterior samples for each effect were used to estimate the effect (e.g., probit differences between treatments) and its 95% credible interval (CrI) and to obtain the rank probability of a treatment being the best or better than each comparator. The estimate of the response probability of achieving each threshold (e.g., PASI 90) for a treatment was similarly obtained by summarizing the corresponding samples of probability. For easier interpretation, the risk ratio and risk difference at each threshold were calculated similarly. The number needed to treat (NNT) for each treatment vs placebo was estimated as the reciprocal of the corresponding treatment’s risk difference with placebo at each threshold. In the binomial analysis, we only estimated the odds ratios and the corresponding CrIs. We only reported the findings of PASI 75, PASI 90 and PASI 100 here although PASI 50 (threshold not of clinical relevance) data were used in our analytical models to improve the model stability given that PASI responses in the placebo arm tended to be low or completely absent with increasing PASI thresholds.
The methods to detect network inconsistency were based on NICE Technical Support Documents, which recommended the use of an unrelated mean effect model [37]. Residual deviance in each arm in each study was also obtained in the multinomial model (for which average deviance over all PASI responses was computed) to evaluate absolute fit to the data. No substantive examples of inconsistency or heterogeneity were detected from investigation of arm-level deviances from the different models that were investigated (badly fitting data contribute to high heterogeneity, inconsistency, or both in a network). Below, we use the frequentist terminology ‘statistically significant’ to refer to 95% CrIs that do not include 1.0 (for odds ratios or risk ratios) or 0 (for probit differences).
Bayesian NMAs of multinomial models were conducted in JAGS (version 4.3.0), and binomial NMAs were conducted in OpenBUGS (version 3.2.3).
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Results
SLR Search Results
The SLR searches yielded 4922 unique publications from the electronic literature databases and 107 from other sources. After screening, 354 publications met the inclusion criteria for the comparative efficacy and safety outcomes. Eighty-six of these trials (reported across 80 publications), including 34,476 patients, met the inclusion criteria for the planned efficacy NMAs. The flow of included studies in the SLR and NMA is summarised in Fig. 1.

PRISMA Flow Diagram of Study Inclusion and Exclusion of Clinical Efficacy and Safety. NMA network meta-analysis, OLE open-label extension, PRISMA Preferred Reporting Items for Systematic Reviews and Meta-Analyses, RCT randomised controlled trial, SLR systematic literature review
Study and Patient Characteristics
Among the included studies, 56 reported on phase 3 or phase 3b trials, 10 were phase 2, 3 were phase 2/3, 2 were phase 4 and 15 did not report trial phase. Total sample sizes of included studies ranged from 20 [38] to 1306 [39] patients, with most studies analysing at least 100 patients. All had similar inclusion/exclusion criteria and definitions of PSO severity. Trial populations were generally similar with respect to age and sex. The mean age of participants in the different treatment arms ranged from 38.3 [40] to 55.3 years old [41] and were mostly males. The proportion of patients with comorbid psoriatic arthritis (PsA) ranged from 2.4% [40] to 36.8% [42]. Patients had moderate to severe PSO for an average of 11 [42] to 24 years [41]. Additional details on patient characteristics are presented in Table S6 in the electronic Supplementary Material. Non-biologic and biologic systemic therapies were evaluated in 16 and 76 trials, respectively. IL inhibitors were more commonly investigated biologics (k = 46) compared to TNF-α inhibitors (k = 40) with etanercept and secukinumab being the most commonly studied interventions as they were evaluated in 11 and 13 trials, respectively.
A total of 67 trials were deemed to have a low risk of bias, 14 were rated as having some concerns, and 5 had a high risk of bias. Summary assessments for each domain and the overall risk of bias are presented in Table S7 in the electronic Supplementary Material.
The feasibility assessment demonstrated that the main sources of heterogeneity among included trials were variation in geography, study years, comorbid PsA, time since diagnosis, prior phototherapy or non-biologic treatment use and prior biologic treatment use (Table S6 in the electronic Supplementary Material). Studies varied widely in reported placebo rates (Figure S1 in the electronic Supplementary Material). Accordingly, baseline-risk adjustment model was undertaken to address this heterogeneity across trials. Network diagrams for the base case analysis and the different sensitivity analyses are presented in Fig. 2.

Network diagram for trials reporting PASI outcomes. ACTR acitretin, ADA adalimumab, APR apremilast, BKZ bimekizumab, BRO brodalumab, CSP cyclosporine, CZP certolizumab pegol, DF dimethyl fumarate, ETN etanercept, GUS guselkumab, IL interleukin, IFX infliximab, IXE ixekizumab, MTX methotrexate, PASI Psoriasis Area and Severity Index, PBO placebo, RZB risankizumab, SEC secukinumab, TIL tildrakizumab, TNF tumour necrosis factor, UST ustekinumab
NMA Results
Model Fit and Diagnostics
The best-fitting model for the base case analytic scenario, which included all eligible evidence, was the baseline-unadjusted FE, REZ model. However, given the small differences in the DIC and based on clinical recommendations, the a priori choice of considering the baseline risk-adjusted model (RE REZ) was selected as our base case model. Model fit results are summarised in Table S8 in the electronic Supplementary Material. No substantive evidence of inconsistency or heterogeneity was detected across the different NMAs.
Efficacy of Bimekizumab Relative to Other Biologic Therapies at 10–16 Weeks
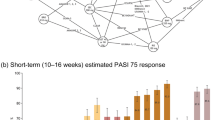
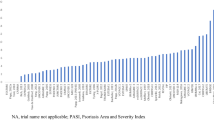
The base case model demonstrated that IL-17 and IL-23 (including bimekizumab 320 mg, risankizumab 150 mg, ixekizumab 80 mg, brodalumab 210 mg, guselkumab 100 mg and secukinumab 300 mg) were the most effective treatments in the network across all PASI response levels. At 10–16 weeks, bimekizumab 320 mg had the highest probability of achieving PASI 75, PASI 90 and PASI 100, with response probabilities of 92.3%, 84.0% and 57.8%, respectively (Fig. 3, and Table S9 in the electronic Supplementary Material). Risankizumab 150 mg and brodalumab 210 mg had the second and third highest probabilities of PASI 90 and PASI 100, respectively.

Probit probabilities (95% CrI) of achieving PASI outcomes (REZ, adjusted, random-effects multinomial model). Treatments are sorted by the highest to lowest probabilities of reaching PASI 75. CrI credible interval, IL interleukin, PASI Psoriasis Area and Severity Index, TNF tumour necrosis factor
Among the TNF-α inhibitors, infliximab 5 mg/kg demonstrated the highest probability of response across all PASI response levels. Both doses of etanercept (25 mg and 50 mg) had the lowest response probabilities among the biologics (Fig. 3). The full results, including non-biologic treatments, can be found in Table S9, Figure S2, and Figure S3 in the electronic Supplementary Material.
Bimekizumab 320 mg was superior in achieving PASI 90 and PASI 100 at the specific thresholds compared with all other treatments in the base case model (Fig. 4). Differences were statistically significant for achieving PASI 90 and PASI 100 compared to all other treatments. For PASI 75, the benefit of bimekizumab was statistically significant compared to all other treatments except risankizumab 150 mg and ixekizumab 80 mg. Findings of the sensitivity analyses conducted using the binomial models were mostly consistent with the base case results for PASI 75 and PASI 90 (Figure S3).

Risk ratios (95% CrI) of achieving PASI 75, PASI 90 and PASI 100 for bimekizumab 320 mg versus other treatments (REZ, adjusted, random-effects multinomial model). Treatments are sorted by the highest to lowest probabilities of reaching PASI 75. CrI credible interval, IL interleukin, PASI Psoriasis Area and Severity Index, TNF tumour necrosis factor
Bimekizumab had the lowest NNT to achieve PASI 75, PASI 90 and PASI 100 when compared with placebo, followed by risankizumab 150 mg, ixekizumab 80 mg and brodalumab 210 mg (Table S10 in the electronic Supplementary Material). The NNTs (95% CrI) for bimekizumab to achieve PASI 75, PASI 90 and PASI 100 were 1.16 (CrI: 1.12, 1.20), 1.22 (CrI: 1.16, 1.29) and 1.74 (CrI: 1.58, 1.96), respectively.
Results of the best fitting model (i.e., baseline-unadjusted FE, REZ model) were consistent with those obtained using our base case model selected a priori (baseline-adjusted RE, REZ), whereby bimekizumab 320 mg had the highest probability of achieving PASI 75, PASI 90 and PASI 100 (Table S11 in the electronic Supplementary Material) and was superior in achieving the different PASI responses compared with all other treatments.
Discussion
This SLR and NMA assessed the comparative efficacy of bimekizumab and other licensed biologic therapies for the treatment of moderate to severe PSO. These findings suggested that IL-17 and IL-23 inhibitors were the most effective treatments in the network across all PASI response levels.
Interleukin-12/23, -17 and -23 inhibitors, except tildrakizumab and secukinumab 150 mg, had a > 70% probability of achieving PASI 75 after 10–16 weeks of treatment. The superiority of IL inhibitors over TNF-α inhibitors and non-biologics in moderate to severe PSO has been well documented in several NMAs. Armstrong et al. [43] and Sawyer et al. [44] demonstrated that ixekizumab, risankizumab and brodalumab had higher response rates at 10–16 weeks versus TNF-α inhibitors and non-biologics. Wright et al. [45] showed that most NMAs evaluating the efficacy of biologic therapies concluded that IL inhibitors were superior to other available therapies in treatment of moderate to severe PSO. These findings align with the central roles IL-17 and -23 cytokines play in the pathogenesis of PSO [46]. In addition, the differences in the efficacy among IL inhibitors evaluated in our NMA can be attributed to the distinct roles played by the different cytokines in the pathogenesis of PSO. Specifically, IL-23 has been shown to play a key role in activating the Th17 pathway and the downstream production of the IL-17 cytokines such as IL-17A and IL-17F. Thus, biologic therapies targeting IL-23, including risankizumab, guselkumab and tildrakizumab have been shown to achieve high levels of skin clearance in PSO [47,48,49]. However, IL-23 inhibition does not result in a complete blockage of IL-17, which is also produced via non-Th17 cells [50, 51]. The direct downstream inhibition of IL-17A cytokines has been also shown to be a clinically effective strategy, as demonstrated by the therapeutic benefit of secukinumab and ixekizumab in moderate to severe PSO [39, 52]. However, given the abundance of IL-17F in psoriatic lesions [53, 54], it has been postulated that the simultaneous selective inhibition of both IL-17A and IL-17F cytokines may provide an additional benefit compared to inhibiting IL-17A only [18]. This has been corroborated in the phase III BE RADIANT trial that demonstrated a greater clinical benefit for the selective dual IL-17A and IL-17F inhibition with bimekizumab compared to IL-17A inhibition only with secukinumab [21].
A key finding of this NMA was the superiority of bimekizumab 320 mg in achieving PASI 90 and PASI 100 at 10 to 16 weeks compared with all treatment options and superiority in achieving PASI 75 at 10 to 16 weeks compared with all treatment options, except ixekizumab 80 mg and risankizumab 150 mg. These findings were consistent when tested using the available binomial RE models, whereby bimekizumab had significantly higher odds of achieving PASI 75 and PASI 90 response levels versus all comparators (except PASI 75 versus risankizumab 150 mg). The magnitude of difference consistently increased with higher PASI responses. Bimekizumab, with its selective inhibition of both IL-17A and IL-17F, offered the additional benefit in achieving complete skin clearance within 10 to 16 weeks of treatment. While Sbidian et al. [55] found that bimekizumab ranked fourth behind infliximab, ixekizumab and risankizumab in achieving PASI 90, their NMA included only a single RCT assessing the efficacy of bimekizumab (BE ABLE 1, a phase 2 dose-finding study, with only 40 patients receiving the licensed dose strength of 320 mg) [19], and estimates of relative effects between treatments were more imprecise than in the current study, which used data from four bimekizumab phase 3/3b trials (BE READY, BE SURE, BE VIVID, and BE RADIANT) [25,26,27,28]. They also included trials investigating non-approved therapies and assessed PASI within a wider time frame (8 to 24 weeks). Although these methodologic differences limit direct comparability, our findings showed that with the inclusion of additional evidence, bimekizumab had a substantial advantage in achieving higher PASI response rates compared with other biologic and non-biologic therapies at 10 to 16 weeks.
The results of our NMA were consistent with the recently published NMA conducted by Armstrong et al. [43] and Shear et al. [56]. Both NMAs demonstrated that biologic treatments were superior over non-biologic treatments in improving short-term PASI outcomes. Among biologics, both NMAs showed that IL inhibitors had better efficacy compared with TNF inhibitors. However, unlike our NMA, none of the aforementioned NMAs included trials that evaluated the efficacy of bimekizumab in PSO. The scope of this NMA differed from several other previously published NMAs; for example, some evaluated unlicensed dosages of available therapies (Jabbar-Lopez et al. [57] and Sbidian et al. [55]) restricted analysis to comparisons of specific treatment classes (Bai et al. [58] and Xu et al. [59]), included paediatric patients (Jabbar-Lopez et al. [57]) or had a different follow-up time (Xu et al. [59] and Sbidian et al. [55]). Our NMA employed distinct methodologic approaches at the level of evidence generation, which aimed to provide clinically meaningful comparative effect estimates among the biologics. For example, the protocol was limited to approved treatments with licensed dosages and restricted the follow-up period (10–16 weeks), which facilitated the interpretation of results in a healthcare decision-making setting.
This study employed the REZ model—an enhancement to the standard multinomial analysis model—to study the comparative efficacy of non-biologic and biologic interventions in achieving improvement in PASI outcomes [35]. The REZ model addresses the inherent drawbacks of both the binomial and standard multinomial models. For instance, in a binomial model, treatments with missing data for certain thresholds are excluded entirely from the corresponding NMA, and these models do not account for the dependence between the ordinal PASI thresholds. In addition, for PASI thresholds with very rare events, binomial models often fail to converge. While these drawbacks are addressed in standard multinomial models, which allow “borrowed strength” across PASI levels, they also assume that treatments have the exact same relative efficacy versus one another for each PASI level. The enhanced REZ multinomial model simultaneously addresses the drawbacks of both the binomial and standard multinomial models by adding a random effects component that allows for variability in relative effects across PASI levels, while still ‘borrowing strength’ through modelling a mean effect between each threshold. Fahrbach et al. [35] demonstrated that the REZ model was associated with considerably better model fit for both the FE versus RE in another NMA conducted in patients with moderate to severe PSO. A multinomial model was employed because of the advantage given by such a model of synthesizing data across dependent outcomes (e.g., PASI 50/75/90/100) simultaneously. Wright et al. [60] concluded that although the choice of multinomial versus binomial models minimally impacted the efficacy and safety estimates, they led to different results at the level of relative ranking of therapies. Baseline risk was simultaneously adjusted for, where possible. Although the unadjusted models were associated with slightly better overall fit compared to the adjusted models in our NMA, there are multiple factors that support the notion of adjusting for placebo response. Slight absolute differences in placebo response rates across trials (e.g., 3% vs 6%) greatly impact unadjusted relative treatment effects. The estimate of the slope of baseline risk was highly significant, and previous considerations of response in autoimmune disorders have also found that adjustment for baseline risk is useful [34].
The strengths of this review include its rigorous methodology, which is adherent to Preferred Reporting Items for Systematic Reviews and Meta-Analyses (PRISMA) guidelines, and its thorough assessment and inclusion of all eligible evidence base. However, our study had some limitations. This review only captured publications available in English and published before July 2020. As a result, more recent, relevant publications may not have been included. Nevertheless, each of the IL inhibitors was represented in at least five trials in the base case network (except for tildrakizumab, which was assessed in three trials). While the inclusion of newer evidence will further improve the robustness of the NMA estimates, it is unlikely to have a substantial impact on our SLR and NMA conclusions. While no major differences in the patient characteristics were found across included trials, a degree of heterogeneity was evident in the prevalence of PsA, time since PSO diagnosis and previous use of systemic therapies. However, the adjustment of baseline risk via placebo rates adjusted for some of the potential heterogeneity in patient characteristics across the trials. Finally, our NMA only evaluated short-term PASI responses and did not investigate the comparative safety among the different treatments, as the assessment of safety end points requires longer term studies that follow substantially higher number of patients.
Conclusions
This NMA demonstrated that IL-17 and IL-23 inhibitors were highly effective in short-term improvement of PSO symptoms among patients with moderate to severe disease. With bimekizumab and its selective inhibition of both IL-17A and IL-17F, patients were significantly more likely to achieve PASI 90 or PASI 100 within 10–16 weeks of the first injection than with all other biologics. Patients with moderate to severe PSO receive lifelong therapy; therefore, future studies should evaluate the risk–benefit balance of available therapies by studying the long-term effectiveness and safety data in open-label extensions and in real-world settings.
References
Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet. 2007;370(9583):263–71.
Gottlieb A, Korman NJ, Gordon KB, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 2. Psoriatic arthritis: overview and guidelines of care for treatment with an emphasis on the biologics. J Am Acad Dermatol. 2008;58(5):851–64.
Parisi R, Iskandar IYK, Kontopantelis E, et al. National, regional, and worldwide epidemiology of psoriasis: systematic analysis and modelling study. BMJ. 2020;369: m1590.
Menter A, Gottlieb A, Feldman SR, et al. Guidelines of care for the management of psoriasis and psoriatic arthritis: Section 1. Overview of psoriasis and guidelines of care for the treatment of psoriasis with biologics. J Am Acad Dermatol. 2008;58(5):826–50.
Mrowietz U, Kragballe K, Reich K, et al. Definition of treatment goals for moderate to severe psoriasis: a European consensus. Arch Dermatol Res. 2011;303(1):1–10.
Gelfand JM, Troxel AB, Lewis JD, et al. The risk of mortality in patients with psoriasis: results from a population-based study. Arch Dermatol. 2007;143(12):1493–9.
Smith CH, Anstey AV, Barker JN, et al. British association of dermatologists’ guidelines for biologic interventions for psoriasis 2009. Br J Dermatol. 2009;161(5):987–1019.
Menter A, Gelfand JM, Connor C, et al. Joint American academy of dermatology-national psoriasis foundation guidelines of care for the management of psoriasis with systemic nonbiologic therapies. J Am Acad Dermatol. 2020;82(6):1445–86.
Smith CH, Yiu ZZN, Bale T, et al. British association of dermatologists guidelines for biologic therapy for psoriasis 2020: a rapid update. Br J Dermatol. 2020;183(4):628–37.
Balak DMW, Gerdes S, Parodi A, Salgado-Boquete L. Long-term safety of oral systemic therapies for psoriasis: a comprehensive review of the literature. Dermatol Therapy. 2020;10(4):589–613.
Menter ASB, Kaplan DH, et al. Joint AAD-NPF guidelines of care for the management and treatment of psoriasis with biologics. J Am Acad Dermatol. 2019;80(4):P1029–72.
Ronholt K, Iversen L. Old and New Biological Therapies for Psoriasis. Int J Mol Sci. 2017;18(11).
Electronic Medicines Compendium. Summary of Product Characteristics: Bimzelx 160 mg solution for injection in pre-filled syringe 2021 [Available from: https://www.medicines.org.uk/emc/product/12833.
European Medicines Agency. Summary of Product Characteristics: Bimzelx 160 mg solution for injection in pre-filled syringe, Bimzelx 160 mg solution for injection in pre-filled pen 2021. https://www.ema.europa.eu/en/documents/product-information/bimzelx-epar-product-information_en.pdf.
BIMZELX[®] (bimekizumab) Approved in Japan for the Treatment of Plaque Psoriasis, Generalized Pustular Psoriasis and Psoriatic Erythroderma [press release]. 24 January 2022.
Government of Canada. Regulatory Decision Summary—Bimzelx—Health Canada 2022. https://hpr-rps.hres.ca/reg-content/regulatory-decision-summary-detail.php?lang=en&linkID=RDS00918.
Australian Government Department of Health. Bimzelx bimekizumab 160 mg/1 mL solution for injection safety syringe 2022. https://www.ebs.tga.gov.au/servlet/xmlmillr6?dbid=ebs/PublicHTML/pdfStore.nsf&docid=FE76410329A0F0FECA25880F003CFB4F&agid=(PrintDetailsPublic)&actionid=1.
Adams R, Maroof A, Baker T, et al. Bimekizumab, a Novel Humanized IgG1 Antibody That Neutralizes Both IL-17A and IL-17F. Front Immunol. 2020;11:1894.
Papp KA, Merola JF, Gottlieb AB, et al. Dual neutralization of both interleukin 17A and interleukin 17F with bimekizumab in patients with psoriasis: Results from BE ABLE 1, a 12-week randomized, double-blinded, placebo-controlled phase 2b trial. J Am Acad Dermatol. 2018;79(2):277–86 e10.
Reich K, Papp KA, Blauvelt A, et al. Bimekizumab versus ustekinumab for the treatment of moderate to severe plaque psoriasis (BE VIVID): efficacy and safety from a 52-week, multicentre, double-blind, active comparator and placebo controlled phase 3 trial. Lancet. 2021;397(10273):487–98.
Reich K, Warren RB, Lebwohl M, et al. Bimekizumab versus Secukinumab in Plaque Psoriasis. N Engl J Med. 2021;385(2):142–52.
Warren RB, Blauvelt A, Bagel J, et al. Bimekizumab versus Adalimumab in Plaque Psoriasis. N Engl J Med. 2021;385(2):130–41.
Higgins JPTTJ, Chandler J, Cumpston M, Li T, Page MJ, Welch VA, editors. Cochrane Handbook for Systematic Reviews of Interventions. 2nd ed. Chichester (UK): John Wiley & Sons; 2019.
Moher D, Liberati A, Tetzlaff J, Altman DG, Group P. Preferred reporting items for systematic reviews and meta-analyses: the PRISMA statement. BMJ. 2009;339:b2535
UCB. Protocol PS0008 Amendment 2: A phase 3, multicenter, randomized, double-blind study with an active-controlled initial treatment period followed by a dose-blind maintenance treatment period to evaluate the efficacy and safety of bimekizumab in adult subjects with moderate to severe chronic plaque psoriasis phase 3. 2018.
UCB. Protocol PS0013 Amendment 3: A phase 3, multicenter, double-blind, placebo-controlled study with an initial treatment period followed by a randomizedwithdrawal period to evaluate the efficacy and safety of bimekizumab in adult subjects with moderate to severe chronic plaque psoriasis phase 3. 2019.
UCB. Protocol PS0009 Amendment 4: A phase 3, multicenter, randomized, double-blind, placebo- and active comparator-controlled, parallel-group study to evaluate the efficacy and safety of bimekizumab in adult subjects with moderate to severe chronic plaque psoriasis phase 3. 2019.
UCB. Protocol PS0015 Amendment 4: A multicenter, randomized, double-blind, secukinumab-controlled, parallel-group study to evaluate the efficacy and safety of bimekizumab in adult subjects with moderate to severe chronic plaque psoriasis phase 3b. 2020.
Sterne JAC, Savovic J, Page MJ, et al. RoB 2: a revised tool for assessing risk of bias in randomised trials. BMJ. 2019;366: l4898.
Salanti G. Indirect and mixed-treatment comparison, network, or multiple-treatments meta-analysis: many names, many benefits, many concerns for the next generation evidence synthesis tool. Res Synth Methods. 2012;3(2):80–97.
Cameron C, Hutton B, Druchok C, et al. Importance of assessing and adjusting for cross-study heterogeneity in network meta-analysis: a case study of psoriasis. J Comp Eff Res. 2018;7(11):1037–51.
Dias S, Sutton A, Welton N, Ades AE. NICE DSU Technical Support Document 3: Heterogeneity: Subgroups, Meta-Regression, Bias and Bias-Adjustment 2011 [updated April 2012. http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD3-Heterogeneity.final-report.08.05.12.pdf.
Dias S, Sutton A, Welton N, Ades AE. NICE DSU Technical Support Document 5: Evidence Synthesis in the Baseline Natural History Model 2012 [updated April 2012. http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD5-Baseline.final-report.08.05.12.pdf.
Dias S, Welton N, Sutton A, Ades AE. NICE DSU Technical Support Document 2: A Generalised Linear Modelling Framework for Pairwise and Network Meta-Analysis of Randomised Controlled Trials 2011 [updated September 2016. http://nicedsu.org.uk/wp-content/uploads/2016/03/A-general-linear-modelling-framework-for-pair-wise-and-network-meta-analysis-of-randomised-controlled-trials..pdf.
Fahrbach K, Sarri G, Phillippo DM, et al. Short-term efficacy of biologic therapies in moderate-to-severe plaque psoriasis: a systematic literature review and an enhanced multinomial network meta-analysis. Dermatol Ther (Heidelb). 2021;11(6):1965–98.
Spiegelhalter D, Best NG, Carlin BP, Van Der Linde A. Bayesian measures of model complexity and fit. J R Stat Soc Stat Methodol Ser B. 2002;64(4):583–639.
Dias S, Welton NJ, Sutton AJ, Caldwell DM, Lu G, Ades AE. NICE DSU Technical Support Document 4: Inconsistency in Networks of Evidence Based on Randomised Controlled Trials 2011 [updated April 2014. http://nicedsu.org.uk/wp-content/uploads/2016/03/TSD4-Inconsistency.final_.15April2014.pdf.
Antiga E, Volpi W, Chiarini C, et al. The role of etanercept on the expression of markers of T helper 17 cells and their precursors in skin lesions of patients with psoriasis vulgaris. Int J Immunopathol Pharmacol. 2010;23(3):767–74.
Langley RG, Elewski BE, Lebwohl M, et al. Secukinumab in plaque psoriasis–results of two phase 3 trials. N Engl J Med. 2014;371(4):326–38.
Heydendael VM, Spuls PI, Opmeer BC, et al. Methotrexate versus cyclosporine in moderate-to-severe chronic plaque psoriasis. N Engl J Med. 2003;349(7):658–65.
Gisondi P, Del Giglio M, Cotena C, Girolomoni G. Combining etanercept and acitretin in the therapy of chronic plaque psoriasis: a 24-week, randomized, controlled, investigator-blinded pilot trial. Br J Dermatol. 2008;158(6):1345–9.
Torii H, Nakagawa H, Japanese Infliximab Study i. Infliximab monotherapy in Japanese patients with moderate-to-severe plaque psoriasis and psoriatic arthritis. A randomized, double-blind, placebo-controlled multicenter trial. J Dermatol Sci. 2010;59(1):40–9.
Armstrong AW, Soliman AM, Betts KA, et al. Comparative efficacy and relative ranking of biologics and oral therapies for moderate-to-severe plaque psoriasis: a network meta-analysis. Dermatol Ther (Heidelb). 2021;11(3):885–905.
Sawyer LM, Malottki K, Sabry-Grant C, et al. Assessing the relative efficacy of interleukin-17 and interleukin-23 targeted treatments for moderate-to-severe plaque psoriasis: a systematic review and network meta-analysis of PASI response. PLoS ONE. 2019;14(8): e0220868.
Wright E, Yasmeen N, Malottki K, et al. Assessing the quality and coherence of network meta-analyses of biologics in plaque psoriasis: What does all this evidence synthesis tell us? Dermatol Therapy. 2021;11(1):181–220.
Chiricozzi A, Romanelli P, Volpe E, Borsellino G, Romanelli M. Scanning the Immunopathogenesis of Psoriasis. Int J Mol Sci. 2018;19(1).
Gordon KB, Strober B, Lebwohl M, et al. Efficacy and safety of risankizumab in moderate-to-severe plaque psoriasis (UltIMMa-1 and UltIMMa-2): results from two double-blind, randomised, placebo-controlled and ustekinumab-controlled phase 3 trials. Lancet. 2018;392(10148):650–61.
Blauvelt A, Papp KA, Griffiths CE, et al. Efficacy and safety of guselkumab, an anti-interleukin-23 monoclonal antibody, compared with adalimumab for the continuous treatment of patients with moderate to severe psoriasis: Results from the phase III, double-blinded, placebo- and active comparator-controlled VOYAGE 1 trial. J Am Acad Dermatol. 2017;76(3):405–17.
Reich K, Papp KA, Blauvelt A, et al. Tildrakizumab versus placebo or etanercept for chronic plaque psoriasis (reSURFACE 1 and reSURFACE 2): results from two randomised controlled, phase 3 trials. Lancet. 2017;390(10091):276–88.
Jones SA, Sutton CE, Cua D, Mills KH. Therapeutic potential of targeting IL-17. Nat Immunol. 2012;13(11):1022–5.
Armstrong AW, Read C, Leonardi C, Kircik L. IL-23 Versus IL-17 in the pathogenesis of psoriasis: there is more to the story than IL-17A. J Drugs Dermatol. 2019;18(8):S202–8.
Gordon KB, Blauvelt A, Papp KA, et al. Phase 3 trials of ixekizumab in moderate-to-severe plaque psoriasis. N Engl J Med. 2016;375(4):345–56.
Kolbinger F, Loesche C, Valentin MA, et al. beta-Defensin 2 is a responsive biomarker of IL-17A-driven skin pathology in patients with psoriasis. J Allergy Clin Immunol. 2017;139(3):923–32 e8.
Johansen C, Usher PA, Kjellerup RB, Lundsgaard D, Iversen L, Kragballe K. Characterization of the interleukin-17 isoforms and receptors in lesional psoriatic skin. Br J Dermatol. 2009;160(2):319–24.
Sbidian E, Chaimani A, Afach S, et al. Systemic pharmacological treatments for chronic plaque psoriasis: a network meta-analysis. Cochrane Database Syst Rev. 2020;1:CD011535.
Shear NH, Betts KA, Soliman AM, et al. Comparative safety and benefit-risk profile of biologics and oral treatment for moderate-to-severe plaque psoriasis: a network meta-analysis of clinical trial data. J Am Acad Dermatol. 2021;85(3):572–81.
Jabbar-Lopez ZK, Yiu ZZN, Ward V, et al. Quantitative evaluation of biologic therapy options for psoriasis: a systematic review and network meta-analysis. J Invest Dermatol. 2017;137(8):1646–54.
Bai F, Li GG, Liu Q, Niu X, Li R, Ma H. Short-Term efficacy and safety of IL-17, IL-12/23, and IL-23 inhibitors brodalumab, secukinumab, ixekizumab, ustekinumab, guselkumab, tildrakizumab, and risankizumab for the treatment of moderate to severe plaque psoriasis: a systematic review and network meta-analysis of randomized controlled trials. J Immunol Res. 2019;2019:2546161.
Xu S, Zhang X, Pan M, Shuai Z, Xu S, Pan F. Treatment of plaque psoriasis with IL-23p19 blockers: a systematic review and meta-analysis. Int Immunopharmacol. 2019;75: 105841.
Wright E, Yasmeen N, Malottki K, et al. Assessing the quality and coherence of network meta-analyses of biologics in plaque psoriasis: what does all this evidence synthesis tell us? Dermatol Ther (Heidelb). 2021;11(1):181–220.
Acknowledgements
Funding
This study and the Rapid Service Fee was funded by UCB Pharma. Richard B. Warren is supported by the Manchester NIHR Biomedical Research Centre.
Medical Writing, Editorial, and Other Assistance
Medical writing and editorial assistance in the preparation of this article was provided by Sean Smith, Colleen Dumont, and Lauren Randall from Evidera and was funded by UCB Pharma. Samantha Martel and Grammati Sarri contributed to the design and execution of the original systematic literature review and meta-analysis, upon which this work is based. The authors also acknowledge Susanne Wiegratz from UCB Pharma for publication coordination.
Authorship
All named authors meet the International Committee of Medical Journal Editors (ICMJE) criteria for authorship for this article, take responsibility for the integrity of the work as a whole, and have given their approval for this version to be published.
Author Contributions
Substantial contributions to study conception and design: KF, MB, BN; substantial contributions to analysis and interpretation of the data: KF, MB, BN, AF, PK, SK, MS, VT; drafting the article or revising it critically for important intellectual content: KF, MB, BN, AF, PK, SK, MS, VT, AA, RW, CM, CL, MA; final approval of the version of the article to be published: KF, MB, BN, AF, PK, SK, MS, VT, AA, RW, CM, CL, MA.
Disclosures
April Armstrong: Has served as a research investigator and/or scientific advisor to AbbVie, Almirall, Arcutis, ASLAN, BI, Bristol Myers Squibb, EPI, Incyte, LEO Pharma, UCB Pharma, Janssen, Lilly, Nimbus, Novartis, Ortho Dermatologics, Sun Pharma, Dermavant, Dermira, Sanofi, Regeneron, and Pfizer. Kyle Fahrbach, Marissa Betts, Binod Neupane, Paulina Kazmierska, Mahmoud Slim: Employees by Evidera, Inc., by PPD. Craig Leonardi: Speaker (honoraria) for AbbVie, Celgene, Eli Lilly and Novartis; served as an investigator for AbbVie, Actavis, Amgen, Boehringer Ingelheim, Celgene, Coherus, Corrona, Dermira, Eli Lilly, Galderma, Glenmark, Janssen, LEO Pharma, Merck (MSD), Novartis, Novella, Pfizer, Sandoz, Stiefel and Wyeth; served on scientific advisory boards for AbbVie, Amgen, Boehringer Ingelheim, Dermira, Eli Lilly, Janssen, LEO Pharma, Pfizer, Sandoz, UCB Pharma and Vitae. President of the International Psoriasis Council Fellow of the American Academy of Dermatology Member of the American Dermatological Association Adjunct Professor of Dermatology at St. Louis University School of Medicine. Private practice in St. Louis, MO. Matthias Augustin: Consulting fees from AbbVie, Almirall, Amgen, Biogen, Boehringer Ingelheim, Celgene, Centocor, Eli Lilly, GSK, Hexal, Janssen, LEO Pharma, Medac, Merck, MSD, Mundipharma, Novartis, Pfizer, Sandoz, UCB Pharma, and Xenoport. Andreas Freitag: Employee of Evidera at the time the research was conducted. AF is currently an employee at Cytel. Sandeep Kiri: Employee of UCB Pharma, stockholder of GSK and UCB Pharma. Vanessa Taieb: Employee of UCB Pharma. Natalie Nunez Gomez: Employee and shareholder of UCB Pharma. Richard B. Warren: Consulting fees from AbbVie, Almirall, Amgen, Arena, Astellas, Avillion, Biogen, Bristol Myers Squibb, Boehringer Ingelheim, Celgene, Eli Lilly, GSK, Janssen, LEO Pharma, Novartis, Pfizer, Sanofi, and UCB Pharma; research grants to his institution from AbbVie, Almirall, Janssen, LEO Pharma, Novartis, and UCB Pharma; honoraria from Astellas, DiCE, GSK, and Union.
Compliance with Ethics Guidelines
This article is based on previously conducted studies and does not contain any new studies with human participants or animals performed by any of the authors.
Data Availability
The datasets generated during and/or analysed during the current study are available from the corresponding author on reasonable request.
Author information
Authors and Affiliations
Corresponding author
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution-NonCommercial 4.0 International License, which permits any non-commercial use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by-nc/4.0/.
About this article

Cite this article
Armstrong, A., Fahrbach, K., Leonardi, C. et al. Efficacy of Bimekizumab and Other Biologics in Moderate to Severe Plaque Psoriasis: A Systematic Literature Review and a Network Meta-Analysis. Dermatol Ther (Heidelb) 12, 1777–1792 (2022). https://doi.org/10.1007/s13555-022-00760-8
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13555-022-00760-8